Pompe disease at a glance
- Affects about one in 58,000 Americans
- Caused by mutation in the GAA gene
- Characterized by a toxic buildup of glycogen
Pompe disease symptoms
- Respiratory issues such as respiratory insufficiency (a condition in which the lungs cannot take in sufficient oxygen or expel sufficient carbon dioxide), chronic respiratory infections, sleep apnea or the need for artificial ventilation
- Muscular disorders such as proximal myopathy (improper functioning of muscle fibers, leading to weakness in muscles close to the torso); progressive muscle weakness in the diaphragm, trunk and lower limbs; wheelchair dependence
- Loss of neuromuscular control, cognitive impairment, reduction in executive function
- Difficulty chewing and swallowing, gastrointestinal symptoms including those resembling irritable bowel disease
- Significantly shortened lifespan
Treating Pompe disease
Pompe disease is treated with enzyme replacement therapy, or ERT.
ERT slows but does not halt the overall progression of disease. In people living with Pompe disease, the body starts to break down the acid alpha-glucosidase (GAA) enzyme immediately after administration of ERT, so people on ERT typically receive lifelong biweekly infusions.
Even on the standard of care, people with Pompe disease may have a significantly shortened life expectancy, depending on the age of onset and the speed of progression, and may experience debilitating symptoms that reduce their quality of life.1,2,3
At AVROBIO, we are developing a new approach that could halt or reverse disease with a single dose. Our approach is designed to circulate the crucial enzyme into the brain as well as cardiac and skeletal muscles. Reaching these tissues and organs is challenging for any treatment, but it is essential to address the symptoms of Pompe.
Our gene therapy program for Pompe is in the preclinical research phase.
What causes Pompe disease?
Pompe disease is caused by a defect in a single gene, known as GAA.
The faulty GAA gene results in a functional deficiency of an enzyme called acid alpha-glucosidase (GAA). That enzyme is essential to breaking down a complex sugar (glycogen) into a simple sugar (glucose) that is needed to fuel cells.4
The lack of this enzyme causes glycogen to accumulate in skeletal and cardiac tissues, as well as in the nervous system, leading to a wide range of symptoms including progressive weakness and loss of motor function.5
There are several forms of Pompe disease. The infantile-onset form of the disease affects infants and is associated with rapid disease progression and a high mortality rate. Late-onset Pompe disease is less aggressive, but still progressive.
People with Pompe disease have a significantly shorter life expectancy than the general population.
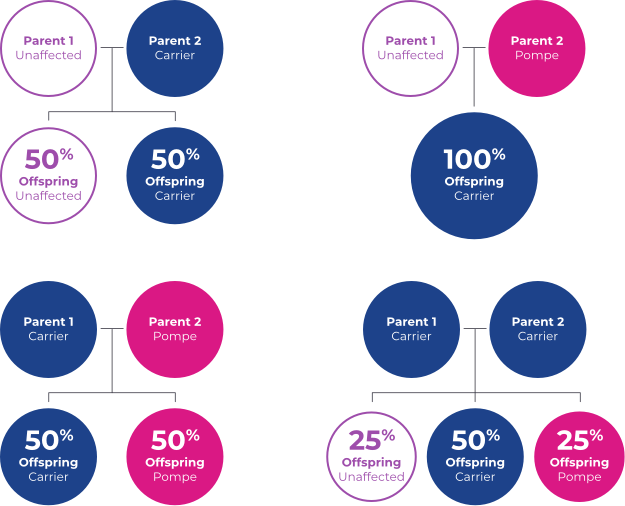
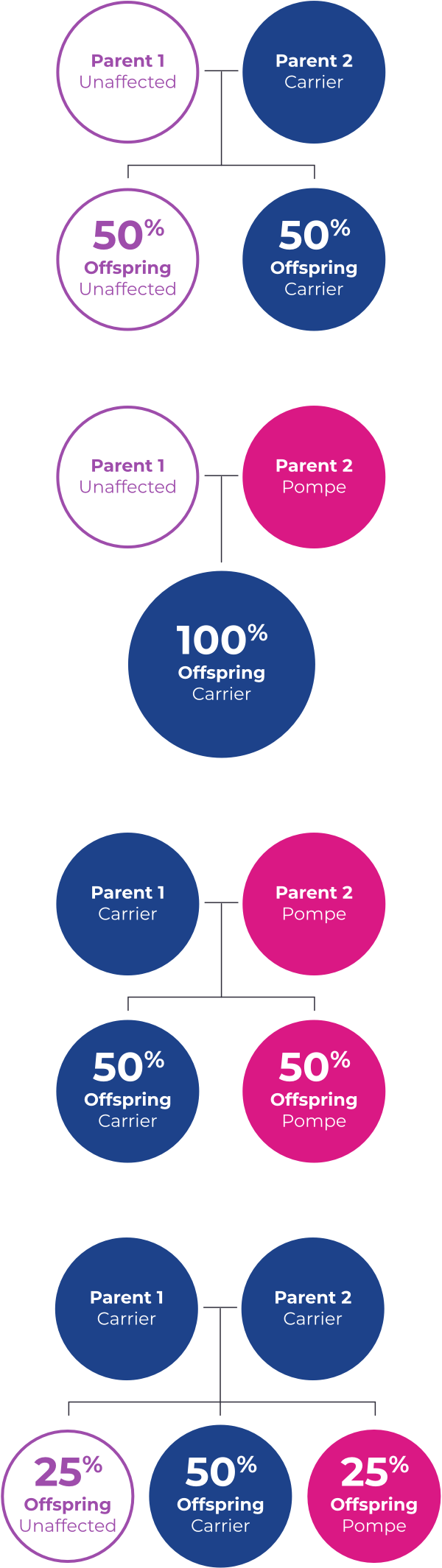
How is Pompe disease inherited?
Healthy people have two working copies of the GAA gene responsible for expressing the enzyme that breaks down glycogen into glucose, which can then be used as fuel for the cell.
When one of those copies is faulty, the individual is considered a “carrier” for Pompe disease but will not develop it. When a carrier has a child, he or she has a 50/50 chance of passing the faulty gene to the child.
A child who inherits two faulty genes may develop Pompe disease. Males and females are equally affected.
*These links are provided for informational purposes only. AVROBIO is not affiliated with any of these institutions and is not responsible for any of the content on their websites.
*AVROBIO may from time to time provide funding and/or support to organizations listed above.
Learn about our research in Pompe disease
We’re leveraging our gene therapy platform to research potential treatments that could halt disease progression or reverse symptoms caused by Pompe disease. Our gene therapy program for Pompe is still in the preclinical research phase.
- Barba-Romero MA, Barrot E, Bautista-Lorite J, Gutiérrez-Rivas E, Illa I, Jiménez LM, et al. Clinical guidelines for late-onset Pompe disease. Rev Neurol 2012; 54: 497-507.
- National Center for Advancing Translational Services (NIH). Glycogen storage disease type 2. Accessed May 2020. https://rarediseases.info.nih.gov/diseases/5714/glycogen-storage-disease-type-2/cases/22926.
- Pompe Disease News. Late-onset Pompe disease. Accessed May 2020. https://pompediseasenews.com/late-onset-pompe-disease/.
- Kishnani PS, et al. Pompe disease diagnosis and management guideline. Genetics in Medicine. 2006;8(5):267-288. doi:10.1097/01.gim.0000218152.87434.f3.
- Hirschhorn R, et al. Glycogen Storage Disease Type II: Acid Alpha-glucosidase (Acid Maltase) Deficiency. In: Scriver C, Beaudet A, Sly W, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. 8th Edition. New York: McGraw-Hill, 3389-3420 (2001).
- Hagemans, M & Janssens, A. Cecile J.W. & Winkel, Léon & Sieradzan, Kasia & Reuser, Arnold & Doorn, Pieter & Ploeg, A. Late-onset Pompe disease primarily affects quality of life in physical health domains. Neurology. 63. 1688-92. 10.1212/01.WNL.0000142597.69707.78. (2004).
- Case LE, et al. Infantile Pompe disease on ERT: update on clinical presentation, musculoskeletal management, and exercise considerations. Am J Med Genet C Semin Med Genet. 2012;160C:69–79.
- National Organization for Rare Diseases (NORD) rarediseases.org/Pompedisease. Accessed August 2017.
- Kemper AR. The Condition Review Workgroup. Evidence Report: Newborn Screening for Pompe Disease (2013). http://www.hrsa.gov/advisorycommittees/mchbadvisory/heritabledisorders/nominatecondition/reviews/pompereport2013.pdf. Accessed August 2017.
AVROBIO
Explore more



Vector Space Blog
Dive into the latest science on gene therapy with our Vector Space Blog.