Hunter syndrome at a glance
- Also known as mucopolysaccharidosis type II (MPS II)
- X-linked recessive genetic condition affects one in 100,000 to one in 170,000 males worldwide1
- Children typically diagnosed between ages 2-6
- Caused by a deficiency of the lysosomal enzyme iduronate-2-sulfatase (IDS) resulting from a change in the IDS gene. The enzyme IDS is essential for the breakdown of large sugar molecules called glycosaminoglycans (GAGs, also known as mucopolysaccharides)2
- Characterized by a toxic buildup of GAGs in multiple organ systems3
Hunter syndrome symptoms
- A wide variety of potential symptoms may occur throughout the body due to the buildup of GAGs. Hunter syndrome can lead to neurological complications, including serious cognitive deficits, as well as complications in skeletal and connective tissue and in the respiratory and cardiac systems3
- Distinct skeletal characteristics include changes in facial features (such as enlarged head, broad nose, flared nostrils or protruding tongue), short stature, a short neck and irregularly shaped and widely spaced teeth4
- Connective tissue changes can include thick skin, joint stiffness with associated restriction of movements, and lump-like skin growths5
- Significant neurological complications can occur in some patients, such as seizures or behavior changes6
- Ear, nose and throat changes can make breathing difficult and lead to chronic ear and sinus infections, respiratory infections3 and pneumonia5
- Other potential symptoms can include3:
- Hydrocephalus (fluid buildup in the cavities deep within the brain)
- Cardiac valve disease
- Hepatosplenomegaly (enlarged liver and spleen)
- Inguinal hernias (tissue swells throughout the gut muscles)
- Impaired vision
- Impaired or loss of hearing
- Spinal stenosis (narrowing of the spaces within the spine)
- Carpal tunnel syndrome
- Hoarse voice
Generally, Hunter syndrome consists of two forms, early progressive and slow progressive, previously referred to as severe and mild forms, respectively.3
Approximately two-thirds7 of individuals with Hunter syndrome have a neuronopathic or early progressive form and may experience developmental and neurological decline early in life. This form may also lead to obstructive airway disease and cardiac failure in the teenage years.7 With early progressive Hunter syndrome, there is generally less symptom variability3.
Approximately one-third7 of individuals with Hunter syndrome have a slow progressive form. They generally do not have neurological symptoms, and the rate of progression may be slower. The age of onset, as well as the amount and extent of symptoms in this group, is variable. Individuals with this form generally have minimal intellectual impairment3.
Treating Hunter syndrome
The standard of care for treating Hunter syndrome is enzyme replacement therapy, or ERT, which can delay some complications but does not halt overall progression of the disease and has not been demonstrated to address cognitive issues. Even with ERT, people with Hunter syndrome face life-limiting symptoms and a significantly reduced life span1.
The enzyme delivered via ERT is metabolized quickly, so people on ERT typically require lifelong weekly infusions8.
At AVROBIO, we are developing a new approach that could prevent the development even in the most severe symptoms of Hunter syndrome with a single dose. Our gene therapy program for Hunter syndrome is in the preclinical research phase.
What causes Hunter syndrome?
Hunter syndrome is caused by a change in a single gene known as IDS.
This mutation results in the inability to produce the functional lysosomal enzyme, iduronate-2-sulfatase (IDS), which is required to break down molecules called GAGs (also known as mucopolysaccharides), which then accumulate throughout the body. This accumulation in tissues and organs leads to numerous symptoms throughout the body including the brain.
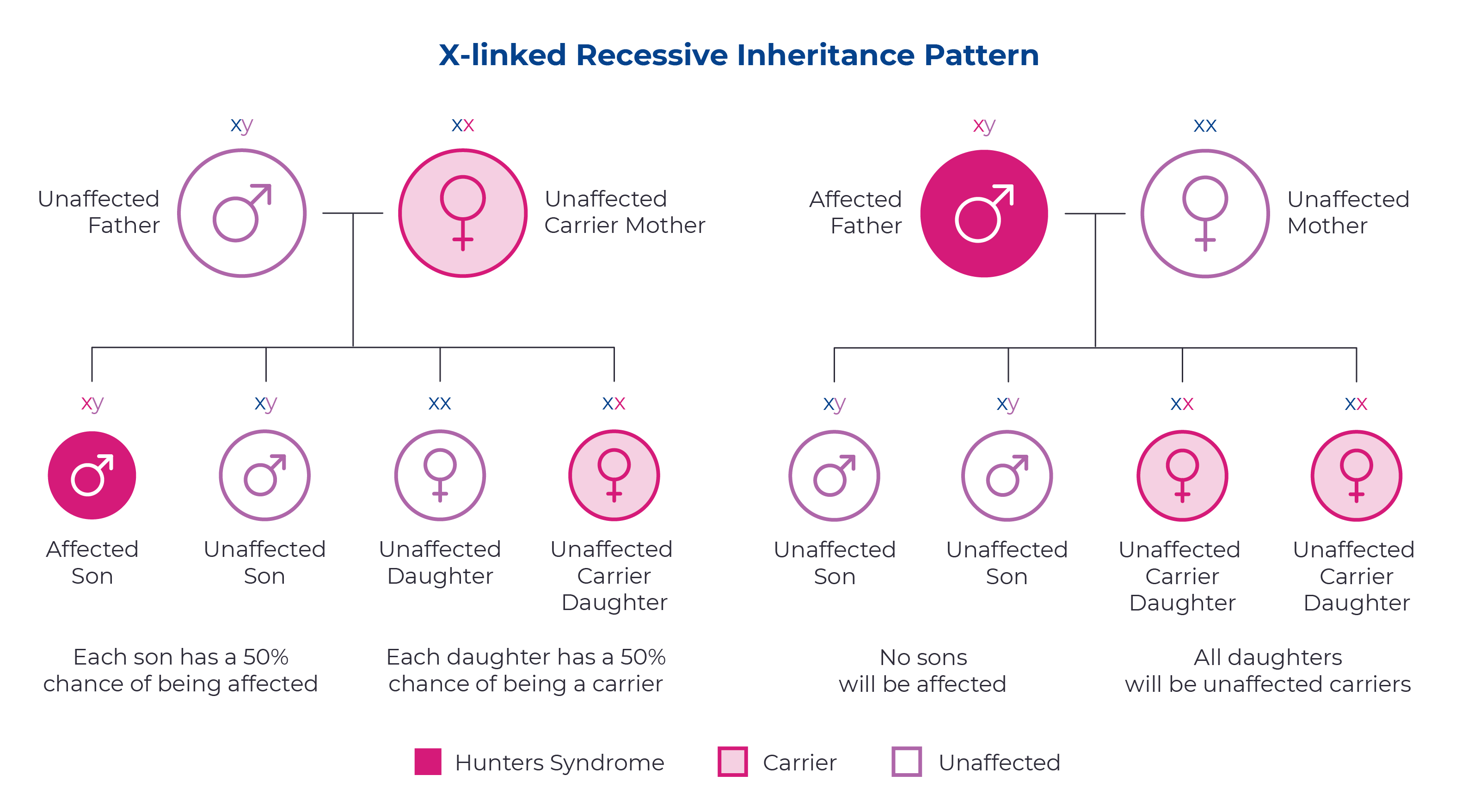
How is Hunter syndrome inherited?
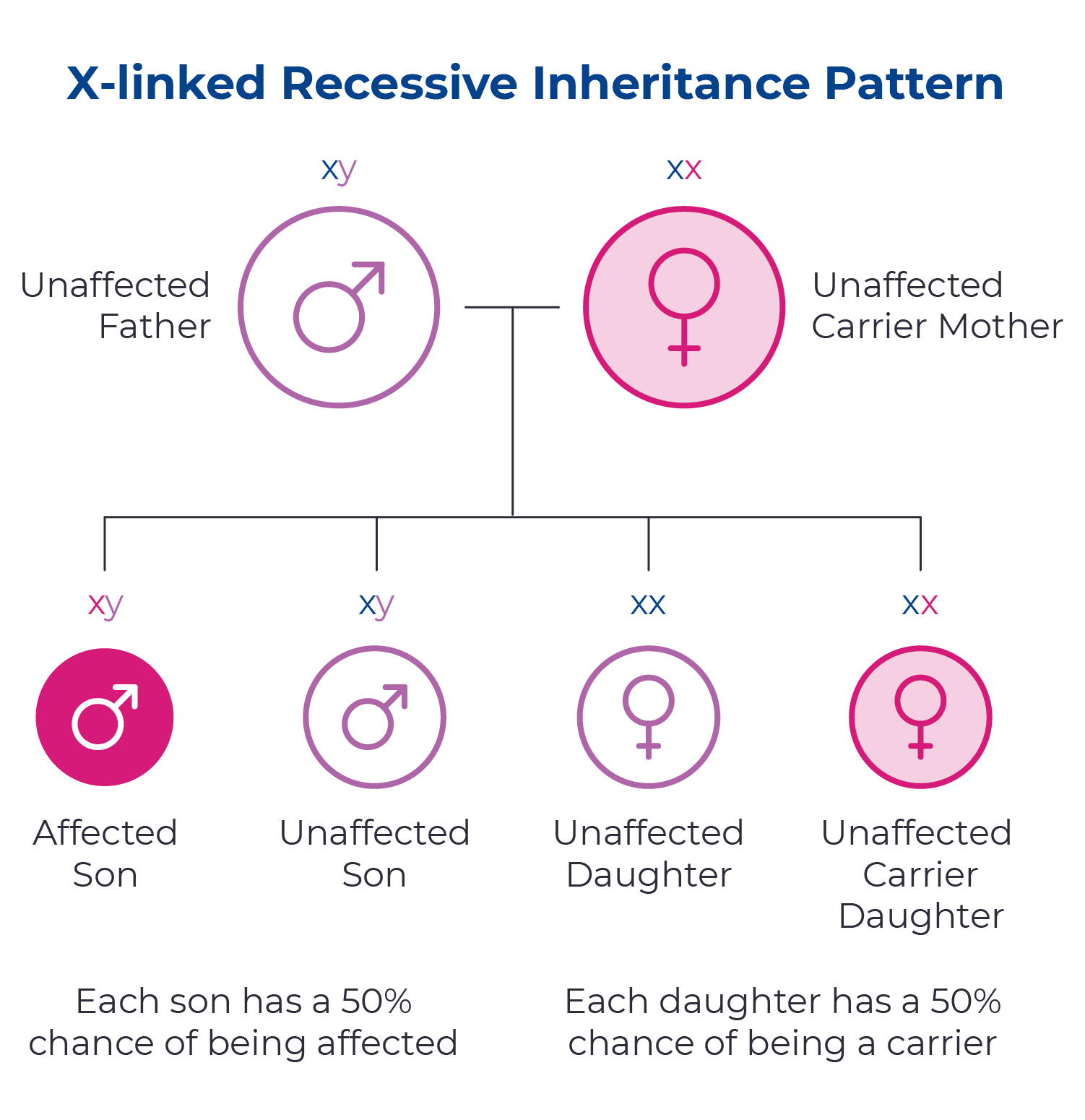
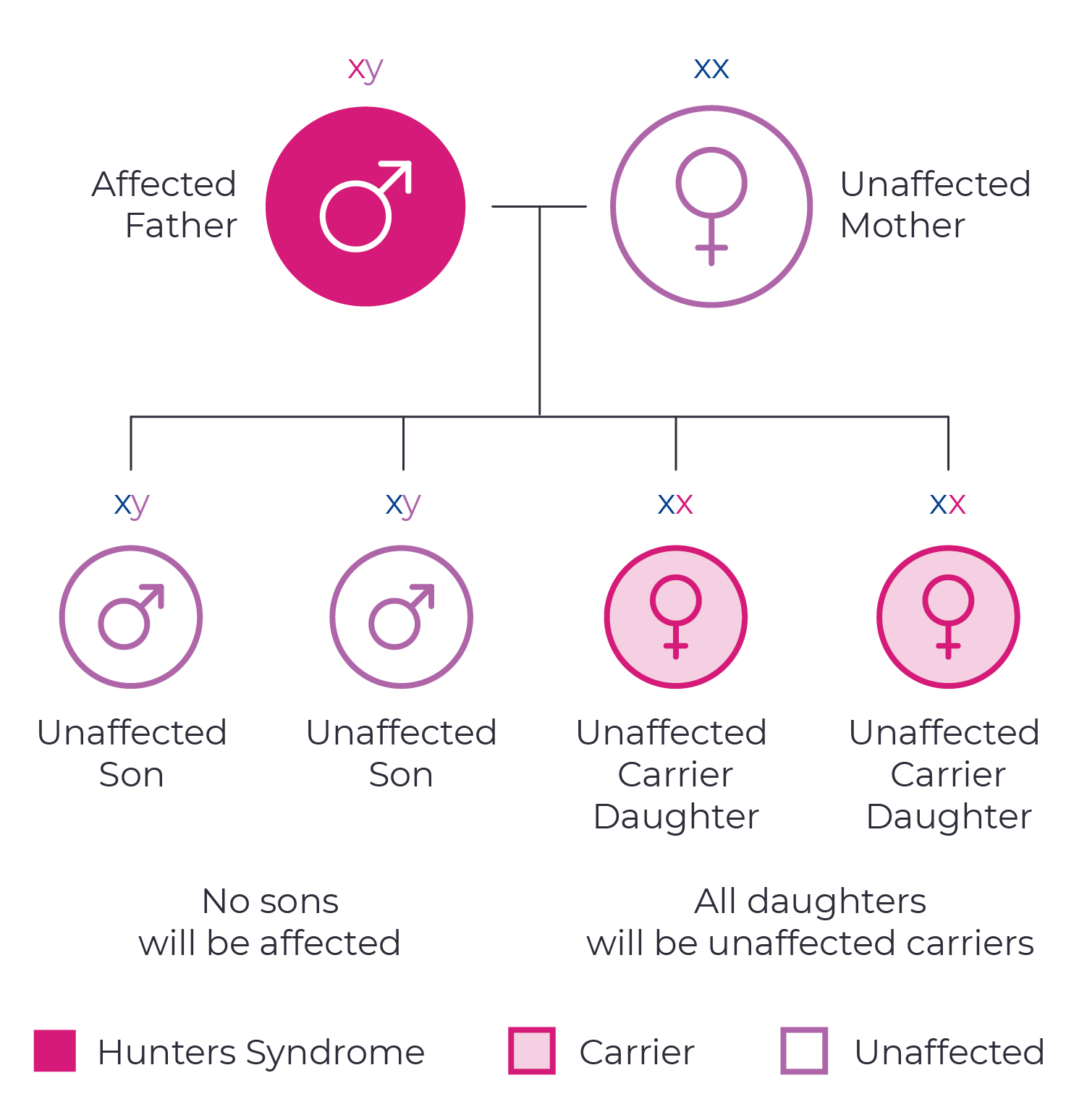
Hunter syndrome is a genetic condition caused by changes in the IDS gene. Since this gene is located on the X chromosome, it is passed down through an X-linked inheritance pattern. This means that mothers with a single copy of the IDS gene have a 50/50 chance of passing on the mutated gene to each of their children. Fathers affected with Hunter syndrome will pass the mutated gene to their daughters, but not their sons.
*These links are provided for informational purposes only. AVROBIO is not affiliated with any of these institutions and is not responsible for any of the content on their websites.
*AVROBIO may from time to time provide funding and/or support to organizations listed above.
Learn about our clinical trial in Hunter syndrome
AVROBIO’s investigational gene therapy for neuronopathic mucopolysaccharidosis type II (nMPSII or Hunter syndrome), is being studied in a Phase 1/2 clinical trial sponsored by the University of Manchester, U.K.
- Mucopolysaccharidosis Type II – Genetics Home Reference – NIH. https://ghr.nlm.nih.gov/condition/mucopolysaccharidosis-type-ii#statistics
- Khan, S. A., et al. Epidemiology of mucopolysaccharidoses. Molecular genetics and metabolism, 121(3), 227–240. (2017). https://doi.org/10.1016/j.ymgme.2017.05.016
- Scarpa M. Mucopolysaccharidosis Type II. 2007 Nov 6 [Updated 2018 Oct. 4]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews®. https://www.ncbi.nlm.nih.gov/books/NBK1274/
- Hunter syndrome. Mayo Clinic. https://www.mayoclinic.org/diseases-conditions/hunter-syndrome/symptoms-causes/syc-20350706
- Mucopolysaccharidoses Fact Sheet, NINDS. NIH Publication No. 19-NS-5115. November 2019. https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Mucopolysaccharidoses-Fact-Sheet
- J.B. Eisengart, et al. The nature and impact of neurobehavioral symptoms in neuronopathic Hunter syndrome. Molecular Genetics and Metabolism Reports, Volume 22, 2020, 100549, ISSN 2214-4269. http://www.sciencedirect.com/science/article/pii/S2214426919301521
- Wraith, J. E., et al. Mucopolysaccharidosis type II (Hunter syndrome): a clinical review and recommendations for treatment in the era of enzyme replacement therapy. European journal of pediatrics, 167(3), 267–277. (2008). https://doi.org/10.1007/s00431-007-0635-4
- Elaprase (idursulfase) injection label. Retrieved June 15, 2020 from https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/125151s0152lbl.pdf
- Mucopolysaccharidosis Type II – NORD. https://rarediseases.org/rare-diseases/mucopolysaccharidosis-type-ii-2/
- Tylki-Szymańska A. Mucopolysaccharidosis type II, Hunter’s syndrome. Pediatr Endocrinol Rev. 2014;12 Suppl 1:107-113. https://pubmed.ncbi.nlm.nih.gov/25345092/
AVROBIO
Explore more


