Patients and FamiliesLysosomal Disorders
ONE LABEL, many different diseases
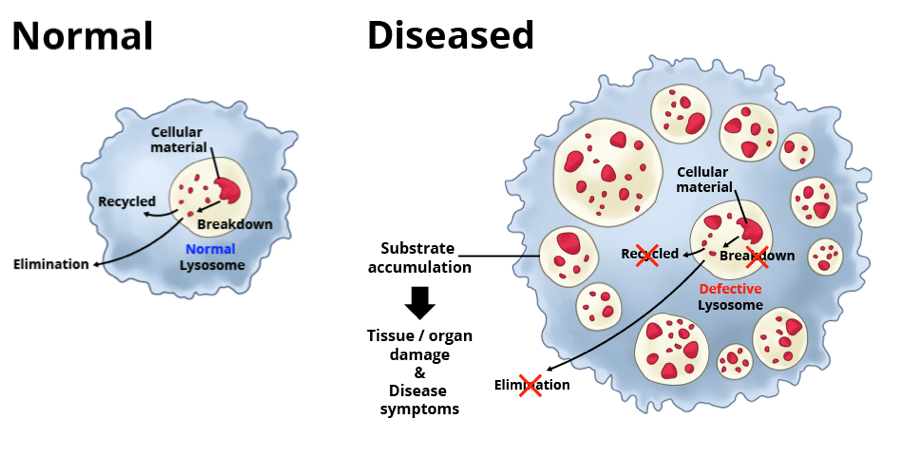
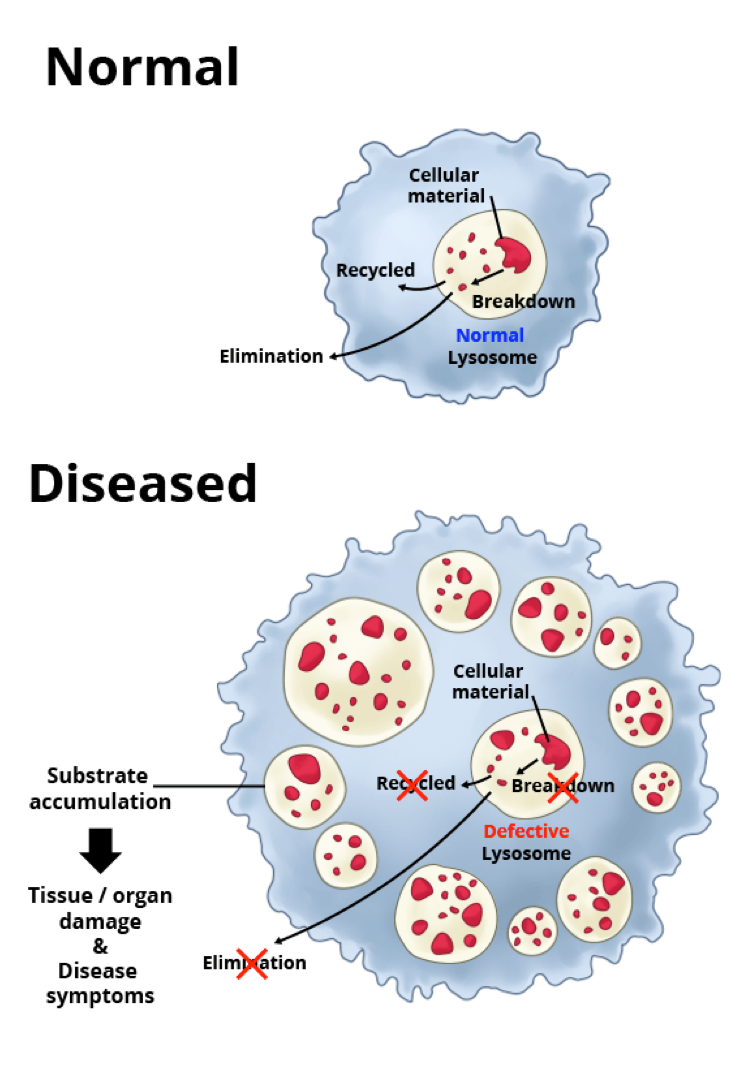
Lysosomal disorders, sometimes called lysosomal storage disorders, are rare diseases caused by a mutation in a single gene. Because of this mutation, the gene fails to make or makes a defective version of, a specific protein that is needed for proper functioning of the lysosomes — important structures within cells that recycle cellular materials. When the lysosomes don’t function properly, toxic materials build up. This, in turn, causes problems for the cell, leading to tissue damage and debilitating symptoms affecting many parts of the body.